5.2 Molecular Dynamics at Constant Temperature
Conventional MD simulation conserves total energy; hence, the time averages computed from MD simulation, if it is long enough, are equivalent to ensemble averages computed from the microcanonical ensemble. The flexibility of MD is greatly enhanced by noting that it is not restricted to NVE. There exist techniques by which MD can simulate in the NVT or NPT ensembles as well. We will consider some popular temperature control schemes, and one popular pressure control scheme, in this section.
There are essentially three ways to control the temperature in an MD simulation:
- Scaling velocities (e.g. simple velocity scaling and the Berendsen thermostat)
- Adding stochastic forces and/or velocities (e.g., the Andersen, Langevin, and Dissipative Particle Dynamics thermostats)
- Using “extended Lagrangian” formalisms (e.g., the Nosé-Hoover thermostat)
Each of these classes of schemes has advantages and disadvantages, depending on the application. In the following subsections, we consider several examples of thermostats, and attempt to discuss their advantages and drawbacks. A simple barostat is also described in the last section.
5.2.1 Velocity Scaling: Isokinetics and the Berendsen Thermostat
First, velocity scaling schemes do not strictly follow the canonical ensemble, though in practice, the amount they deviate from canonical is quite small. (This can be measured by comparing the velocity distribution function with a Gaussian.) It is relatively easy to implement the second class of schemes, because they can be “dropped” in to existing codes using almost any integrator. However, they suffer the drawback that they are not time-reversible or deterministic, properties that become important in some advanced MD techniques. The third class are slightly more difficult to implement, but do not suffer from such drawbacks as time-irreversibility.
We have in effect already encountered simple velocity scaling in mdlj.c, in the initialization function. Here, particle velocities are chosen randomly from \([-0.5,0.5]\) and the rescaled to result in a desired temperature given by the relation: \begin{equation} \label {eq:mdens1} \frac {3}{2}Nk_BT = \frac {1}{2}\sum _im_iv_i^2 \end{equation} We could, if we wanted to, turn this into a dynamic scheme for continually keeping the velocities scaled such that the total kinetic energy is constant. We can measure the instaneous temperature immediately after a velocity update, and call it \(T_i\). Eq. 150 indicates that if we scale velocities by a constant \(\lambda \), where \begin{equation} \lambda = \sqrt {\left (T/T_i\right )} \end{equation} we will be left with a system at temperature \(T\). Velocity scaling to maintain constant \(\mathscr {K}\) is called an isokinetic thermostat. Such a thermostat cannot be used to conduct a simulation in the canonical ensemble, but is perfectly fine to use in a warmup or initialization phase. We could perform velocity rescaling at every step, or only every few steps. As a suggested exercise, modify mdlj.c to perform velocity scaling to a user-specified setpoint temperature every \(m\) time steps, where \(m\) is a user-defined interval between velocity scaling events. Begin with system at \(T\) = 1.0, and command it to jump to \(T\) = 3.0 after 1,000 steps. How does the system behave, and is it sensitive to your choice of \(m\)?
Another popular velocity scaling thermostat is that of Berendsen [10]. Here, the scale factor is given by \begin{equation} \lambda = \left [1+\frac {\Delta t}{\tau _T}\left (\frac {T}{T_0} - 1\right )\right ]^{\frac {1}{2}} \end{equation} Here, \(T_0\) is the setpoint temperature, \(\Delta t\) is the integration time step, and \(\tau _T\) is a constant called the “rise time” of the thermostat. It describes the strength of the coupling of the system to a hypothetical heat bath. The larger \(\tau _T\), the weaker the coupling; in other words, the larger \(\tau _T\), the longer it takes to achieve a given \(T_0\) after an instantaneous change from some previous \(T_0\). The code mdlj_ber.c implements the Berendsen thermostat. As a brief exercise, you can experiment with this code to get a feeling for how various values of the rise time affect the response of the system when the setpoint temperature is changed instantaneously from 1.0 to 2.0. Below is a lin-log plot of just such an experiment with \(N\) = 256 particles at a density of 0.5. Each curve corresponds to a different value of \(\tau _T\), and they increase by factors of 10. The corresponding time at which the setpoint \(T\) is reached is also seen to increase by the same factor.
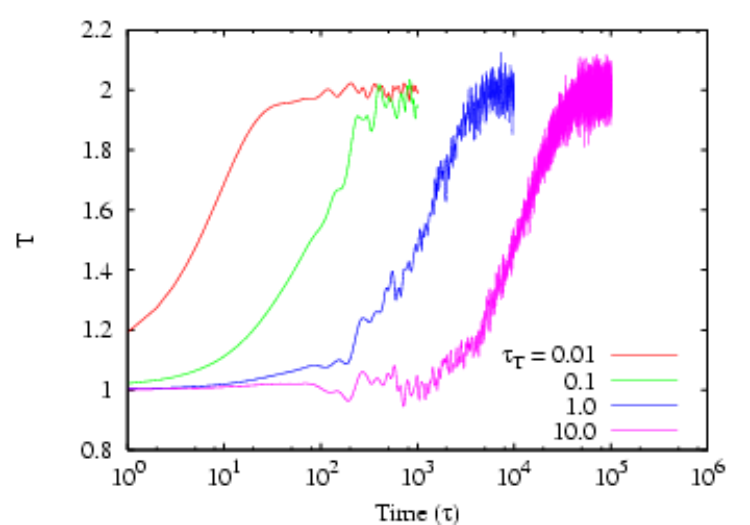
Though relatively simple, velocity scaling thermostats are not recommended for use in production MD runs because they do not strictly conform to the canonical ensemble.
5.2.2 Stochastic NVT Thermostats: Andersen, Langevin, and Dissipative Particle Dynamics
The Andersen Scheme. Perhaps the simplest thermostat which does correctly sample the NVT ensemble is due to Andersen [11]. Here, at each step, some prescribed number of particles is selected, and their momenta (actually, their velocities) are drawn from a Gaussian distribution at the prescribed temperature: \begin{equation} \label {eq:ens:gaus} \mathscr {P}(p) = \left (\frac {\beta }{2\pi m}\right )^{3/2} \exp \left [-\beta p^2/\left (2m\right )\right ] \end{equation} This is intended to mimic collisions with bath particles at a specified \(T\). The strength of the coupling to the heat bath is specified by a collision frequency, \(\nu \). For each particle, a random variate is selected between 0 and 1. If this variate is less than \(\nu \Delta t\), then that particle’s momenta are reset.
The code mdlj_and.c implements the Andersen thermostat with the velocity Verlet algorithm for the Lennard-Jones fluid. I was curious about the importance placed on the fact that a Gaussian distribution of velocities is established during an MD simulation using the Andersen thermostat. F&S seem to indicate that the truth of this fact means that we are sampling the canonical ensemble. But they do not show data for the Berendsen thermostat, for which we should not sample NVT. The figure below is a comparison of the measured velocity distribution for a 20,000-step run at \(T\) = 1.0 and \(\rho = 0.8442\) using the Berendsen thermostat with \(\tau = 0.1\) and a similar run with the Andersen thermostat with \(\nu = 0.01\). Apparently, the Berendsen thermostat also reproduces the correct distribution.
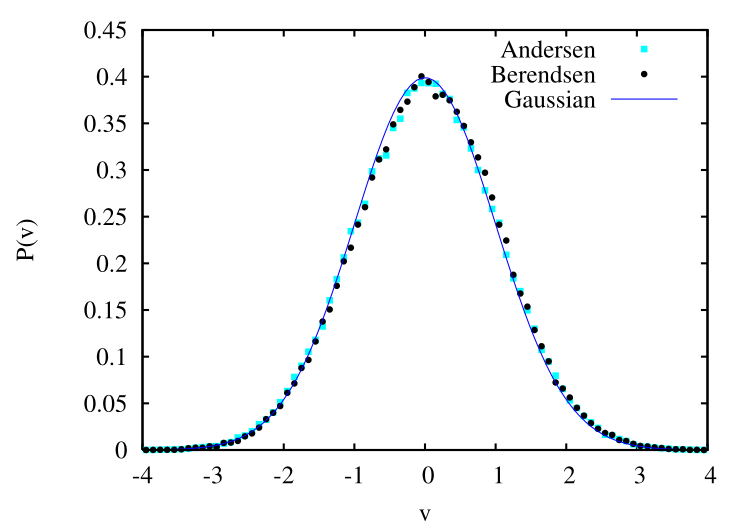
Although they may not be on the right track with regard to the velocity distributions, F&S do make a very good point with regard to measuring transport properties from NVT MD simulations. The Andersen thermostat destroys momentum transport because of the random velocities; hence, there is no continuity of momentum in an Andersen LJ fluid, and therefore no proper \(\mathscr {D}\) or viscosity. The data they cite in Fig. 6.3 clearly shows that \(\mathscr {D}\), if measured from an Andersen MD run, is incorrect.
The Langevin thermostat. In the “Langevin” thermostat, at each time step all particles receive a random force and have their velocities lowered using a constant friction. [12] The average magnitude of the random forces and the friction are related in a particular way, which guarantees that the “fluctuation-dissipation” theorem is obeyed, thereby guaranteeing NVT statistics.
In this formalism, the particle-\(i\) equation of motion is modified: \begin{equation} \label {ens:md:lang} m\ddot {\bf r}_i = -\nabla _i U - m\Gamma \dot {\bf r}_i + {\bf W}_i\left (t\right ) \end{equation} Here, \(\Gamma \) is a friction coefficient with units of \(\tau ^{-1}\), and \({\bf W}_i\) is a random force that is uncorrelated in time and across particles, with a mean given by \begin{equation} \left \langle {\bf W}_i\left (t\right ),{\bf W}_j\left (t^\prime \right )\right \rangle = \delta _{ij}\delta \left (t-t^\prime \right )6k_BmT\Gamma \end{equation}
The code mdlj_lan.c implements the Langevin thermostat. The two major elements are a force initialization at each time step that adds in the random forces, \(\bf W\), and a slight modification to the update equations in the integrator to include the effect of \(\Gamma \). Note that the initialization of forces in the force routine has been removed.
One advantage of the Langevin thermostat (and to a limited extent, the Andersen thermostat and other stochastic-based thermostats) is that we can get away with a larger time step than in NVE simulations. At a density of \(\rho \) = 0.8442 and a mean temperature \(T\) = 1.0, an NVE simulation is unstable for time-steps above about \(\Delta t\) = 0.004. We can, however, run a Langevin dynamics simulation with a friction \(\Gamma \) = 1.0 stably with a time-step as large as \(\Delta t\) = 0.01 or even higher. This has proven invaluable in simulations of more complicated systems that simple liquids, namely linear polymers, which have very long relaxation times. MD with the Langevin thermostat is the method of choice for equilibrating samples of liquids of long bead-spring polymer chains.
Of course, the drawback of most stochastic thermostats (one exception is discussed next) is that momentum transfer is destroyed. So again, it is unadvisable to use Langeving or Andersen thermostats for runs in which you wish to compute diffusion coefficients. I echo the recommendation of F&S: when possible, use NVE to compute properties, and use thermostats for equilibration only.
The Dissipative Particle Dynamics thermostat. The DPD thermostat [13, 14] adds pairwise random and dissipative forces to all particles, and has been shown to preserve momentum transport. Hence, it is the only stochastic thermostat so far that should even be considered for use if one wishes to compute transport properties.
The DPD thermostat is implemented by slight modification of the force routine to add in the pairwise random and dissipative forces. For the \(ij\) pair, the dissipative force is defined as \begin{equation} {\bf f}_{ij}^D = -\gamma \omega ^D\left (r_{ij}\right )\left ({\bf v}_{ij}\cdot \hat {\bf r}_{ij}\right )\hat {\bf r}_{ij} \end{equation} Here, \(\gamma \) is a friction coefficient, \(\omega \) is a cut-off function for the force as a function of the scalar distance between \(i\) and \(j\) which simply limits the interaction range of the dissipative (and random) forces, \({\bf v}_{ij} = {\bf v}_i - {\bf v}_j\) is the relative velocity of \(i\) to \(j\), and \(\hat {\bf r}_{ij} = {\bf r}_{ij}/r_{ij}\) is the unit vector pointing from \(j\) to \(i\). The random force is defined as \begin{equation} {\bf f}_{ij}^R = \sigma \omega ^R\left (r_{ij}\right )\zeta _{ij}\hat {\bf r}_{ij} \end{equation} Here, \(\sigma \) is the strength of the random force, \(\omega ^R\) is a cut-off, and \(\zeta _{ij}\) is a Gaussian random number with zero mean and unit variance, and \(\zeta _{ij} = \zeta _{ji}\).
The update of velocity uses these new forces: \begin{equation} \label {eq:dpd_update} {\bf v}_i\left (t + \Delta t\right ) = {\bf v}_i\left (t\right ) - \frac {\Delta t}{m}\nabla _iU + \frac {\Delta t}{m}{\bf f}_i^D + \frac {\sqrt {\Delta t}}{m}{\bf f}_i^R \end{equation} where \begin{equation} \begin {aligned} {\bf f}_i^D & = \sum _{j\ne i} {\bf f}_{ij}^D \\ {\bf f}_i^R & = \sum _{j\ne i} {\bf f}_{ij}^R \end {aligned} \end{equation}
The parameters \(\gamma \) and \(\sigma \) are linked by a fluctuation-dissipation theorem: \begin{equation} \sigma ^2 = 2\gamma k_B T \end{equation} So, in practice, one must specify either \(\gamma \) or \(\sigma \), and then a setpoint temperature, \(T\), in order to use the DPD thermostat.
The cutoff functions are also related: \begin{equation} \omega ^D\left (r_{ij}\right ) = \left [\omega ^R\left (r_{ij}\right )\right ]^2 \end{equation} This is the only real constraint on the cutoffs; we are otherwise allowed to use any cutoff we like. The simplest uses the cutoff radius of the pair potential, \(r_c\): \begin{equation} \omega \left (r\right ) = \begin {cases} 1 & r < r_c\\ 0 & r > r_c \end {cases} \end{equation} Note that, with this choice, \(\left [\omega ^R\left (r_{ij}\right )\right ]^2\) = \(\omega ^R\left (r_{ij}\right )\) = \(\omega ^D\left (r_{ij}\right ) = \omega \).
The code mdlj_dpd.c implements the DPD thermostat in an MD simulation of the Lennard-Jones liquid. The major changes (compared to mdlj.c) are to the force routine, which now requires several more arguments, including particle velocities, and parameters for the thermostat. Inside the pair loop, the force on each particle is updated by the conservative, dissipative, and random pairwise force components. The random force is divided by \(\sqrt {\Delta t}\) so that the velocity Verlet algorithm need not be altered to implement Eq. 158.
The behavior of the DPD thermostat can be assessed in a similar fashion as was the Berendsen thermostat above. Below is a lin-log plot of system temperature after the setpoint is changed from 1.0 to 2.0, for a Lennard-Jones system \(N\) = 256 and \(\rho \) = 0.8442. Again, each curve corresponds to a different value of \(\gamma \), and they increase by factors of 10. The corresponding time at which the setpoint \(T\) is reached is also seen to increase by the same factor.
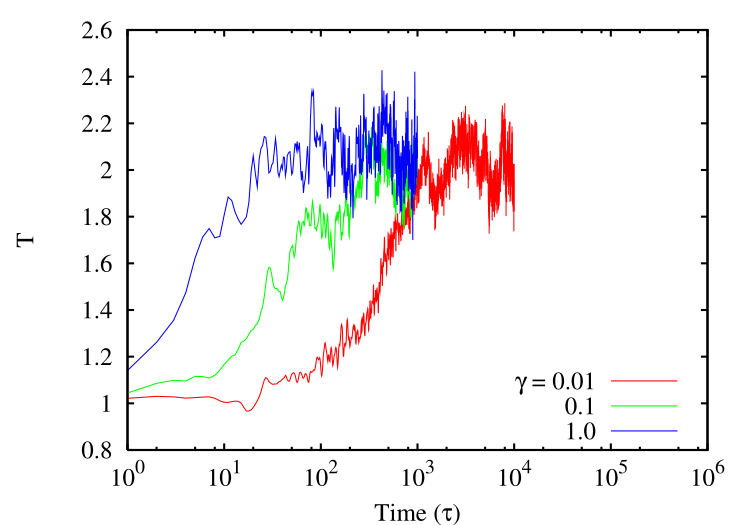
If you have been paying attention, you will notice that in all implementations given here, the thermostat is applied each and every time step. In principle, one could modulate the strength of a thermostat by controlling the frequency with which it is applied. As an exercise, you can pick one of these implementations and modify it such that the thermostat is applied once per \(M\) time steps, where \(M\) is specified on the command-line. You can compare the performance of the thermostat in the same way we have already seen (time required to go from \(T\) = 1.0 to \(T\) = 2.0 upon an instantaneous change in the setpoint \(T\)) for a given friction or strength and for various values of \(M\).
5.2.3 The Nosé-Hoover Chain
The final thermostat we consider is one based on the extended Lagrangian formalism, which leads to a deterministic trajectory; i.e., there are no random forces or velocities to deal with. The most common and so far most reliable thermostat of this kind is the Nosé-Hoover thermostat. This thermostat can be implemented as a “single” or a “chain”; here, we consider a chain.
The basic idea of the Nosé-Hoover thermostat is to use a friction factor to control particle velocities. This friction factor is actually the scaled velocity, \(v_{\xi _1}\), of an additional and dimensionless degree of freedom, \(\xi _1\). This degree of freedom has an associated “mass”, \(Q_1\), which effectively determines the strength of the thermostat. The equations of motion obeyed by this additional degree of freedom guarantee that the original degrees of freedom (\({\bf r}^N\), \({\bf p}^N\)) sample a canonical ensemble. This degree of freedom is the terminus of a chain of similar degrees of freedom, each with their own mass. The chain has a total of \(M\) “links.” The overall set of equations of motion are:
The main advantage of the Nosé-Hoover chain thermostat is that the dynamics of all degrees of freedom are deterministic and time-reversible. No random numbers are used. The code mdlj_nhc.c implements an \(M\) = 2 Nosé-Hoover chain thermostat in an MD simulation of an Lennard-Jones fluid, by implementing Algorithms 30, 31, and 32 from F&S. The two masses of the thermostats are defaulted to \(Q_1\) = \(Q_2\) = 0.1. This “low” mass results in a “loose” control of temperature; higher masses mean a tighter control. \(\xi _1\) is the degree of freedom whose velocity is used to scale particle velocities, so one might hypothesize that the mass \(Q_1\) is the more important. (You can verify this as an exercise.) Though I haven’t verified that my code is 100% bug-free, apparently, the effect of increasing the mass of the coupling degree of freedom is to lengthen the decay time constant of the response to an instantaneous temperature jump:
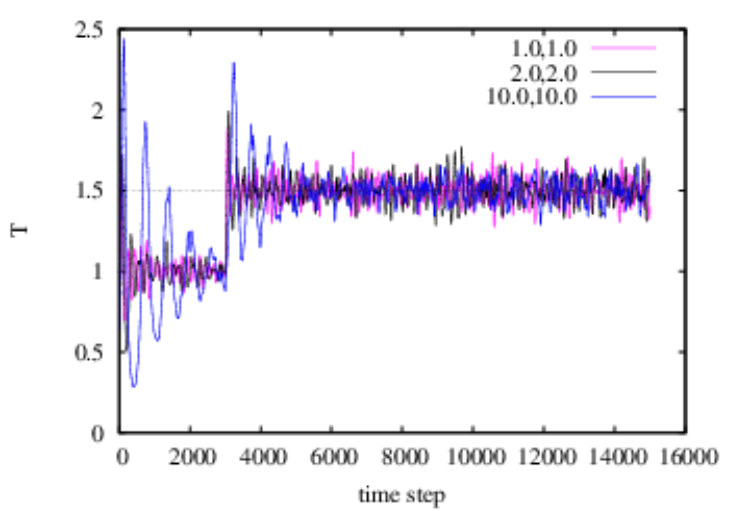
If anyone finds a bug in this code, I will buy you a refreshing beverage of your choice.