7.1 Case Study: Stillinger-Weber Silicon and the Tersoff Potential
No one should have the impression that people who do molecular simulation only care about the Lennard-Jones fluid. It has been and continues to be an important test-bed for theories of the liquid state and phase-equilibria. Nevertheless, molecular simulation has been performed on a wide variety of materials.
As a single example, consider silicon. Perhaps the earliest attempt to use molecular simulation to study a realistic atomic-scale model of silicon was due to Stillinger and Weber [16]. I “cut my teeth” on the Stillinger-Weber potential coding up my first real research code in graduate school. I now make a version of that code available to the students in this course, available at mdswsi.c. This code computes in reduced units as well; \(\sigma \) = 0.20951 nm, \(\epsilon \) = 2.1678 eV, and \(m\) = 28.085 amu, which are appropriate for a system of pure silicon. One reduced unit of temperature, \(T [=] \epsilon /k_B\), corresponds to 25156.73798 K.
The main reason to introduce Stillinger-Weber silicon here is to give you an example of a three-body potential. Silicon forms 4-coordinated tetrahedral bonded structures. The object of the three-body component of the potential is to enforce the tetrahedral bond angle (109.47\(^\circ \)) among triplets of bonded atoms.
The total potential is expressed as two sums, one for unique pair interactions, and another for unique triplet interactions:
The two-body models the bonds: \begin{equation} v_2 \left (r\right ) = \left \{\begin {array}{ll} \epsilon A \left (Br^{-p}-r^{-q}\right )\exp \left [\left (r-a\right )^{-1}\right ], & \mbox {$r < a$}\\ 0 & \mbox {$r\ge a$} \end {array} \right . \end{equation} It is very much like a Lennard-Jones potential, only with different exponents and a “smooth cutoff” as the interatom separation distance, \(r\), approaches some cutoff, \(a\), given by the factor \(\exp \left [\left (r-a\right )^{-1}\right ]\).
The three body models the angles, and is the sum of functions of each of the three angles of a triplet, \(ijk\):
Here I have employed the shorthand notation \(\displaystyle h_{jik} \equiv h\left (r_{ij}, r_{ik}, \theta _{jik}\right )\). Note that, in the notation of this potential, \(\theta _{jik}\) is subtended at \({\bf r}_i\), and \(\cos \theta _{jik}^c = -\frac {1}{3}\):
One computes the angle-\(j\) term, \(h_{ijk}\), and the angle-\(k\) term, \(h_{ikj}\), by permuting the indices appropriately.
The parameters used in the original study by Stillinger and Weber are: \begin{equation} \begin {aligned} A & = 7.049556277 \\ B & = 0.6022245584 \\ p & = 4 \\ q & = 0 \\ a & = 1.80 \\ \lambda & = 21.0 \\ \gamma & = 1.20 \end {aligned} \end{equation}
As in any MD simulation, one computes the force on any particle \(i\) from the negative gradient of the total potential: \begin{equation} \begin {aligned} {\bf f}_i & = -\nabla _{{\bf r}_i}\mathscr {U} \\ & = -\sum _{j\ne i} \nabla _{{\bf r}_i}v_2\left (r_{ij}\right ) - \sum _{j\ne i}\sum _{k\ne i,j} \nabla _{{\bf r}_i}v_3\left ({\bf r}_i,{\bf r}_j,{\bf r}_k\right ) \end {aligned} \end{equation}
The two-body term for the \(i\)-\(j\) interaction is only slightly more complicated than Lennard-Jones, due to the smooth cutoff. Here, assuming \(i\) and \(j\) are within interaction range (\(r_{ij} < a\)), we have for the force on \(i\) due to \(j\): \begin{equation}\label {eq:swfij} \begin {aligned} -\nabla _{{\bf r}_i} v_2\left (r_{ij}\right ) &= -\frac {\partial }{\partial {\bf r}_i} \left \{\epsilon A \left (Br_{ij}^{-p}-r_{ij}^{-q}\right )\exp \left [\left (r_{ij}-a\right )^{-1}\right ]\right \} \\ &= -\epsilon A \frac {{\bf r}_{ij}}{r_{ij}}\left (\left .\left [\frac {\partial }{\partial r} \left (Br^{-p}-r^{-q}\right )\right ]\right |_{r_{ij}}\exp \left [\left (r_{ij}-a\right )^{-1}\right ]\right . \\ &+ \left .\left (Br_{ij}^{-p}-r_{ij}^{-q}\right )\left .\left \{\frac {\partial }{\partial r}\exp \left [\left (r-a\right )^{-1}\right ]\right \}\right |_{r_{ij}}\right ) \\ &= -\epsilon A \frac {{\bf r}_{ij}}{r_{ij}}\left \{\left (-pBr_{ij}^{-p-1}+qr_{ij}^{-q-1}\right )\exp \left [\left (r_{ij}-a\right )^{-1}\right ]\right . \\ &- \left .\left (Br_{ij}^{-p}-r_{ij}^{-q}\right )\exp \left [\left (r_{ij}-a\right )^{-1}\right ]\left (r_{ij}-a\right )^{-2}\right \} \\ &= v_2\frac {{\bf r}_{ij}}{r_{ij}}\left [\frac {pBr_{ij}^{-p-1}-qr_{ij}^{-q-1}}{Br_{ij}^{-p}-r_{ij}^{-q}} + \left (r_{ij}-a\right )^{-2}\right ] \\ &\equiv {\bf f}_{ij} \end {aligned} \end{equation} Note that it is still true that \({\bf f}_{ij} = -{\bf f}_{ji}\). Exercise: Show that the right-hand-side of Eq. 185 is well-behaved (that is, it does not diverge, and in fact vanishes) as \(r_{ij}\rightarrow a^-\).
It is comparatively much more tedious to evaluate the three-body gradients. \begin{equation}\label {eq:sw3_partials} \begin {aligned} -\nabla _{{\bf r}_i}v_3\left ({\bf r}_i,{\bf r}_j,{\bf r}_k\right ) &= -\nabla _{{\bf r}_i}\left (h_{jik} + h_{ijk} + h_{ikj}\right ) \\ &= -\left (\frac {\partial h_{jik}}{\partial {\bf r}_i} +\frac {\partial h_{ijk}}{\partial {\bf r}_i} +\frac {\partial h_{ikj}}{\partial {\bf r}_i}\right ) \\ &\equiv {\bf f}_{i\leftarrow jk} \end {aligned} \end{equation} The triplet forces on the other members of the triplet, \({\bf f}_{j\leftarrow ik}\) and \({\bf f}_{k\leftarrow ij}\), are defined analogously.
Each of the partials in Eq. 186 is unique: \begin{equation} \label {eq:sw3jik} \begin {array}{lll} \mbox {\raisebox {-0.5cm}{\includegraphics [width=1cm]{figpng/hjik.png}}\ \ } \displaystyle {\frac {\partial h_{jik}}{\partial {\bf r}_i}} & \displaystyle = &\displaystyle - \gamma h_{jik}\left [ {{{\bf r}_{ij}}\over {r_{ij}}}{{1}\over {\left (r_{ij}-a\right )^2}} + \frac {{\bf r}_{ik}}{r_{ik}}\frac {1}{\left (r_{ik}-a\right )^2} \right ] \\ & \displaystyle + & \displaystyle 2\lambda \exp \left ( \frac {\gamma }{r_{ij}-a} + \frac {\gamma }{r_{ik}-a} \right ) \left (\cos \theta _{jik}-\cos \theta _{jik}^c\right ) \\ & \displaystyle \times & \displaystyle \left [ \frac {{\bf r}_{ij}}{r_{ij}}\frac {1}{r_{ik}} +\frac {{\bf r}_{ik}}{r_{ik}}\frac {1}{r_{ij}} -\left (\frac {{\bf r}_{ij}}{r_{ij}}\frac {1}{r_{ik}} +\frac {{\bf r}_{ik}}{r_{ik}}\frac {1}{r_{ij}}\right ) \cos \theta _{jik}\right ] \end {array} \end{equation} \begin{equation} \label {eq:sw3ijk} \begin {array}{lll} \mbox {\raisebox {-0.5cm}{\includegraphics [width=1cm]{figpng/hijk.png}}\ \ } \displaystyle \frac {\partial h_{ijk}}{\partial {\bf r}_i} & \displaystyle = & \displaystyle - \gamma _j h_{ijk}\left [ \frac {{\bf r}_{ij}}{r_{ij}}\frac {1}{\left (r_{ij}-a\right )^2} \right ] \\ & \displaystyle + & \displaystyle 2\lambda _j\exp \left ( \frac {\gamma _j}{r_{ij}-a} + \frac {\gamma _j}{r_{jk}-a} \right ) \left (\cos \theta _{ijk}-\cos \theta _{ijk}^c\right ) \\ & \displaystyle \times & \displaystyle \left [ \frac {{\bf r}_{jk}}{r_{jk}}\frac {1}{r_{ij}} +\frac {{\bf r}_{ij}}{r_{ij}}\frac {1}{r_{ij}} \cos \theta _{ijk}\right ] \end {array} \end{equation} \begin{equation} \label {eq:sw3ikj} \begin {array}{lll} \mbox {\raisebox {-0.5cm}{\includegraphics [width=1cm]{figpng/hikj.png}}\ \ } \displaystyle \frac {\partial h_{ikj}}{\partial {\bf r}_i} & \displaystyle = & - \displaystyle \gamma _k h_{ikj}\left [ \frac {{\bf r}_{ik}}{r_{ik}}\frac {1}{\left (r_{ik}-a\right )^2} \right ] \\ & \displaystyle + & \displaystyle 2\lambda _k\exp \left ( \frac {\gamma _k}{r_{ik}-a} + \frac {\gamma _k}{r_{jk}-a} \right ) \left (\cos \theta _{ikj}-\cos \theta _{ikj}^c\right ) \\ & \displaystyle \times & \displaystyle \left [ -\frac {{\bf r}_{jk}}{r_{jk}}\frac {1}{r_{ik}} +\frac {{\bf r}_{ik}}{r_{ik}}\frac {1}{r_{ik}} \cos \theta _{ikj}\right ] \end {array} \end{equation} Note that the right hand sides of each of Eqns. 187, 188, and 189 vanish when the appropriate \(r\)’s are greater than \(a\)’s, as in Eq. 212. Exercise: Show that \({\bf f}_{i\leftarrow jk} + {\bf f}_{j\leftarrow ik} + {\bf f}_{k\leftarrow ij} = 0\).
We can investigate what happens when we willfully ignore the three-body terms. Let us initialize atoms on a diamond-cubic lattice (the minimal energy lattice of silicon); a snapshot appears below.

Let us now run two MD simulations for 10,000 time steps; one with and and the other without three body forces, at reduced initial temperature \(T\) = 0.12 and reduced density \(\rho \) = 0.46 for a small system of 64 atoms. You can download small 64-atom system configuration here, already equilibrated at \(T\)=0.12 (pictured above). Note that you must compile the code mdswsi.c with the flag -DTHREEBODY to include three-body forces. You can create an executable version of mdswsi that ignores three-body forces by compiling without the flag -DTHREEBODY. For example:
% gcc -o mdswsi_no3 mdswsi.c -lm -lgsl % gcc -o mdswsi mdswsi.c -DTHREEBODY -lm -lgsl
Note also that this code uses the Andersen thermostat by default, with a default value of \(\nu \) = 0.1. This can be overridden (or turned off) by specifying a value of \(\nu \) on the command line. For example,
% mdswsi -nu 0 -ns 10001 -icf init.xyz
turns off the thermostat, which is what I have done for this little demonstration.
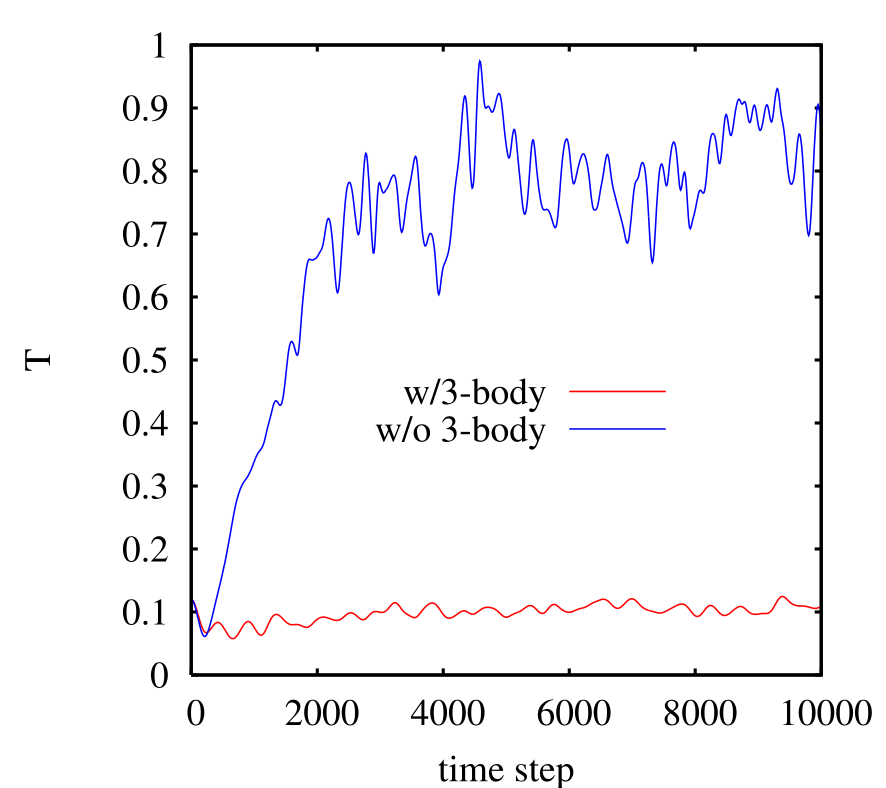
Below we plot the instantaneous temperature vs. time-step (0.001 \(\tau \)) for the two runs. Notice that the system with the 3-body forces intact remains steady at \(T \approx \) 0.12, while the system without 3-body forces simply “melts,” with \(T\) approaching 20,000 K A quick look at a configuration (not shown) reveals that there is no longer any crystalline order; the system is now an amorphous blob of silicon atoms. The lesson of this little demonstration is that one must have three-body forces to stabilize a diamond-cubic lattice.
As a suggested project, you can try one or more of several things with this code.
- The force routine involves a triplet loop, which is extremely costly. Were one to double the size of the system, the computational effort would increase by a factor of eight. Appendix F in F&S discusses algorithms to speed up force evaluation by taking advantage of finite interaction ranges. Can you make the triplet loop more efficient using these techniques? (My thesis code used a unique version of the Verlet List technique.)
- Use the code rdf.c to compute the radial distribution function of both crystalline and liquid silicon. Can you explain the differences between these two, and from the \(g(r)\) for a simple LJ liquid?
- Can you compute the diffusivity of a Si atom in a liquid?